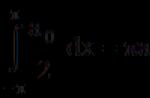Definición 1
La termodinámica estadística es una rama amplia de la física estadística que formula leyes que conectan todas las propiedades moleculares de las sustancias físicas con cantidades medidas durante los experimentos.
Figura 1. Termodinámica estadística de moléculas flexibles. Author24 - intercambio en línea de trabajos de estudiantes
El estudio estadístico de los cuerpos materiales se dedica a fundamentar los postulados y métodos de la termodinámica de los conceptos de equilibrio y al cálculo de funciones importantes utilizando constantes moleculares. La base de esta dirección científica son hipótesis y suposiciones confirmadas por experimentos.
A diferencia de la mecánica clásica, en termodinámica estadística sólo se estudian las lecturas medias de coordenadas y momentos internos, así como la posibilidad de aparición de nuevos valores. Las propiedades termodinámicas de un medio macroscópico se consideran parámetros generales de características o cantidades aleatorias.
Hoy en día, los científicos distinguen entre termodinámica clásica (Boltzmann, Maxwell) y cuántica (Dirac, Fermi, Einstein). La teoría básica de la investigación estadística: existe una relación inequívoca y estable entre las características moleculares de las partículas que forman un sistema particular.
Definición 2
Un conjunto en termodinámica es un número casi infinito de conceptos termodinámicos que se encuentran en microestados diferentes e igualmente probables.
Los parámetros promedio de un elemento observado físicamente durante un largo período de tiempo comienzan a equivaler al valor general del conjunto.
Idea básica de termodinámica estadística.
Figura 2. Formulación estadística de la 2da ley de la termodinámica. Author24 - intercambio en línea de trabajos de estudiantes
La termodinámica estadística establece e implementa la interacción de sistemas microscópicos y macroscópicos. En el primer enfoque científico, basado en la mecánica clásica o cuántica, los estados internos del medio se describen detalladamente en forma de coordenadas y momento de cada partícula individual en un momento determinado. La formulación microscópica requiere resolver ecuaciones de movimiento complejas para muchas variables.
El método macroscópico utilizado por la termodinámica clásica caracteriza exclusivamente el estado externo del sistema y utiliza para ello un pequeño número de variables:
- temperatura del cuerpo físico;
- volumen de elementos que interactúan;
- número de partículas elementales.
Si todas las sustancias están en un estado de equilibrio, entonces sus indicadores macroscópicos serán constantes y sus coeficientes microscópicos cambiarán gradualmente. Esto significa que cada estado en termodinámica estadística corresponde a varios microestados.
Nota 1
La idea principal de la rama de la física que se estudia es la siguiente: si cada posición de los cuerpos físicos corresponde a muchos microestados, entonces cada uno de ellos, como resultado, hace una contribución significativa al macroestado general.
De esta definición cabe destacar las propiedades elementales de la función de distribución estadística:
- normalización;
- certeza positiva;
- el valor promedio de la función de Hamilton.
El promedio de los microestados existentes se lleva a cabo utilizando el concepto de conjunto estadístico ubicado en cualquier microestado correspondiente a un macroestado. El significado de esta función de distribución es que generalmente determina el peso estadístico de cada estado del concepto.
Conceptos básicos en termodinámica estadística.
Para describir estadística y competentemente los sistemas macroscópicos, los científicos utilizan datos de conjuntos y del espacio de fases, lo que les permite resolver problemas clásicos y cuánticos utilizando el método de la teoría de la probabilidad. El conjunto microcanónico de Gibbs se utiliza a menudo para estudiar sistemas aislados con un volumen y un número constantes de partículas cargadas idénticamente. Este método se utiliza para describir cuidadosamente sistemas de volumen estable que están en equilibrio térmico con el medio ambiente con un índice constante de partículas elementales. Los parámetros de estado de un gran conjunto permiten determinar el potencial químico de sustancias materiales. El sistema isobárico-isotermo de Gibbs se utiliza para explicar la interacción de cuerpos que se encuentran en equilibrio térmico y mecánico en un determinado espacio a presión constante.
El espacio de fases en termodinámica estadística caracteriza un espacio mecánico-multidimensional, cuyos ejes son todos coordenadas generalizadas y los impulsos internos asociados de un sistema con grados de libertad constantes. Para un sistema formado por átomos cuyos indicadores corresponden a la coordenada cartesiana, el conjunto de parámetros y energía térmica se designará según el estado inicial. La acción de cada concepto está representada por un punto en el espacio de fase, y el cambio en un macroestado en el tiempo está representado por el movimiento de un punto a lo largo de la trayectoria de una línea específica. Para describir estadísticamente las propiedades del medio ambiente, se introducen los conceptos de función de distribución y volumen de fase, que caracterizan la densidad de probabilidad de encontrar un nuevo punto que represente el estado real del sistema, así como en la materia cerca de una línea con ciertas coordenadas.
Nota 2
En mecánica cuántica, en lugar de volumen de fase, se utiliza el concepto de espectro de energía discreto de un sistema de volumen finito, ya que este proceso no está determinado por coordenadas y momento, sino por una función de onda, que en un estado dinámico corresponde a todo el espectro de estados cuánticos.
La función de distribución del sistema clásico determinará la posibilidad de implementar un microestado específico en un elemento del volumen del medio de fase. La probabilidad de encontrar partículas en un espacio infinitesimal se puede comparar con la integración de elementos sobre las coordenadas y momentos del sistema. El estado de equilibrio termodinámico debe considerarse como un indicador límite de todas las sustancias, donde surgen soluciones a la ecuación de movimiento de las partículas que componen el concepto para la función de distribución. El tipo de funcional, que es el mismo para los sistemas cuánticos y clásicos, fue establecido por primera vez por el físico teórico J. Gibbs.
Cálculo de funciones estadísticas en termodinámica.

Para calcular correctamente la función termodinámica, es necesario aplicar cualquier distribución física: todos los elementos del sistema son equivalentes entre sí y corresponden a diferentes condiciones externas. La distribución microcanónica de Gibbs se utiliza principalmente en estudios teóricos. Para resolver problemas específicos y más complejos se consideran conjuntos que tienen energía con el medio ambiente y pueden intercambiar partículas y energía. Este método es muy conveniente para estudiar equilibrios químicos y de fases.
Las funciones de partición permiten a los científicos determinar con precisión las propiedades energéticas y termodinámicas de un sistema, obtenidas diferenciando indicadores según parámetros relevantes. Todas estas cantidades adquieren significado estadístico. Así, el potencial interno de un cuerpo material se identifica con la energía media del concepto, lo que permite estudiar la primera ley de la termodinámica, como ley básica de conservación de la energía durante el movimiento inestable de los elementos que componen el sistema. . La energía libre está directamente relacionada con la función de partición del sistema, y la entropía está directamente relacionada con el número de microestados en un macroestado particular y, por lo tanto, con su probabilidad.
El significado de entropía, como medida del surgimiento de un nuevo estado, se conserva en relación con un parámetro arbitrario. En un estado de equilibrio completo, la entropía de un sistema aislado tiene un valor máximo en condiciones externas inicialmente especificadas correctamente, es decir, el estado general de equilibrio es un resultado probable con el máximo peso estadístico. Por lo tanto, una transición suave desde una posición de no equilibrio a una de equilibrio es un proceso de cambio hacia un estado más real.
Este es el significado estadístico de la ley de entropía interna creciente, según la cual aumentan los parámetros de un sistema cerrado. En el cero absoluto, cualquier concepto se encuentra en un estado estable. Esta declaración científica representa la tercera ley de la termodinámica. Vale la pena señalar que para una formulación inequívoca de la entropía es necesario utilizar sólo la descripción cuántica, ya que en la estadística clásica este coeficiente se define con la máxima precisión hasta un término arbitrario.
Conferencia 2.
Termodinámica, física estadística, entropía de la información.
1. Información procedente de termodinámica y física estadística. Función de distribución. Teorema de Liouville. Distribución microcanónica. La primera ley de la termodinámica. Procesos adiabáticos. Entropía. Peso estadístico. La fórmula de Boltzmann. Segunda ley de la termodinámica. Procesos reversibles e irreversibles.
2. Entropía de la información de Shannon. Bits, nueces, trits, etc. Relación entre entropía e información.
Esta parte pertenece a la lección 1. Se considera mejor en la sección V (“El concepto de entrelazamiento de estados cuánticos”).
LE CNOT se representa como:
Almacenamos el valor de (qu)bit a mientras (qu)bit b cambia según la ley XOR:
poco b(objetivo = objetivo) cambia su estado si y sólo si el estado del bit de control a coincide con 1; Al mismo tiempo, el estado del bit de control no cambia.
La operación lógica XOR (CNOT) ilustra por qué los datos clásicos se pueden clonar pero los datos cuánticos no. Tenga en cuenta que en el caso general, por datos cuánticos entenderemos superposiciones de la forma
![]() , (1)
, (1)
donde y son números complejos o amplitudes de estados, y, .
Según la tabla de verdad, si se aplica XOR a datos booleanos en los que el segundo bit está en el estado "0" (b) y el primero está en el estado "X" (a), entonces el primer bit no cambia, y el segundo se convierte en su copia:
U XOR (X, 0) = (X, X), donde X = “0” o “1”.
En el caso cuántico, los datos denotados por el símbolo “X” deben considerarse superposición (1):
![]() .
.
Físicamente, los datos se pueden codificar, por ejemplo, en la base de polarización |V> = 1, |H> = 0 (H,V)= (0,1):
![]() Y
Y ![]()
Se puede observar que realmente se produce una copia de estado. El teorema de la no clonación afirma que es imposible copiar arbitrario estado cuántico. En el ejemplo considerado, la copia se produjo porque la operación se realizó en su propia base (|0>, |1>), es decir V privado caso de un estado cuántico.
Parecería que la operación XOR también se puede utilizar para copiar superposiciones de dos estados booleanos, como |45 0 > ? |V> + |H>:
![]()
¡Pero eso no es cierto! La unitaridad de la evolución cuántica requiere que una superposición de estados de entrada se transforme en una superposición correspondiente de estados de salida:
![]() (2)
(2)
Este es el llamado un estado entrelazado (Ф+), en el que cada uno de los dos qubits de salida no tiene un valor específico (en este caso, polarización). Este ejemplo muestra que las operaciones lógicas realizadas en objetos cuánticos siguen reglas diferentes a las de los procesos informáticos clásicos.
Surge la siguiente pregunta: Parece estar en modo de salida A nuevamente se puede representar como una superposición ![]() , como el estado de la moda b. ¿Cómo demostrar que esto no es así, es decir, que no tiene ningún sentido hablar de estados de modo (bits)? a y la moda (un poco) b?
, como el estado de la moda b. ¿Cómo demostrar que esto no es así, es decir, que no tiene ningún sentido hablar de estados de modo (bits)? a y la moda (un poco) b?
Usemos la analogía de la polarización cuando
![]() (3).
(3).
Hay dos maneras. El camino 1 es más largo, pero más consistente. Es necesario calcular los valores promedio de los parámetros de Stokes para ambos modos de salida. Los promedios se toman de la función de onda (2). Si todos excepto resultan ser iguales a cero, entonces este estado no está polarizado, es decir mixto y la superposición (3) no tiene sentido. Trabajamos en la representación de Heisenberg, cuando los operadores se transforman, pero la función de onda no.
Entonces lo encontramos de moda. a.
![]() - intensidad total del haz a,
- intensidad total del haz a,
![]() - proporción de polarización vertical,
- proporción de polarización vertical,
![]() - compartir +45 0ª polarización,
- compartir +45 0ª polarización,
![]() - proporción de polarización circular derecha.
- proporción de polarización circular derecha.
La función de onda sobre la cual se realiza el promedio se toma en la forma (2):
¿Dónde están los operadores de nacimiento y destrucción en mods? a Y b operar de acuerdo con las reglas:

(Haga los cálculos en la Sección V (ver cuaderno). Allí también calcule la probabilidad de registrar coincidencias o un correlacionador de la forma ![]() }
}
¡Path II es más visual, pero menos “honesto”!
Encontremos la dependencia de la intensidad de la luz en el modo. a sobre el ángulo de rotación de la Polaroid colocada en este modo. Esta es una forma óptica cuántica estándar de verificar el estado (2): la intensidad no debe depender de la rotación. Al mismo tiempo, una dependencia similar del número de coincidencias tiene la forma
![]() . Estas dependencias fueron obtenidas por primera vez por E. Fry (1976) y A. Aspek (1985) y a menudo se interpretan como prueba de la no localidad de la mecánica cuántica.
. Estas dependencias fueron obtenidas por primera vez por E. Fry (1976) y A. Aspek (1985) y a menudo se interpretan como prueba de la no localidad de la mecánica cuántica.
Entonces, la situación experimental se muestra en la figura:

Por definición
![]()
¿Dónde está el operador de aniquilación en el modo a? Se sabe que la transformación de los operadores de dos modos x e y polarizados ortogonalmente cuando la luz pasa a través de una polaroid orientada en ángulo tiene la forma:
![]() .
.
(sólo los términos primero, cuarto, quinto y octavo son distintos de cero) =
(solo el primer y octavo término son diferentes de cero) = - ¡¿no depende del ángulo?!
Físicamente esto sucede porque la función de onda (2) no factoriza y no tiene sentido hablar de estados en modos. A Y b por separado. Por lo tanto, ¡no se puede argumentar que el modo a está en un estado de superposición (3)!
Comentario. Los cálculos realizados (Camino II) no prueban en absoluto que el Estado esté de moda. A no polarizado. Por ejemplo, si en este modo hubiera luz polarizada circularmente, el resultado sería el mismo. Prueba rigurosa, por ejemplo, mediante los parámetros de Stokes (en la sección V).
Tenga en cuenta que actuando de la misma manera, podemos demostrar que el estado en modo a antes del elemento CNOT está polarizado.

En este caso se debe promediar la función de onda del estado inicial (3). El resultado se ve así:
![]() aquellos. los recuentos máximos se alcanzan en = 45 0 .
aquellos. los recuentos máximos se alcanzan en = 45 0 .
Información y entropía.
Sin introducir por ahora el término “operativo” “información”, argumentaremos utilizando un lenguaje “cotidiano”. Aquellos. La información es algún conocimiento sobre un objeto.
El siguiente ejemplo muestra que los conceptos de información y entropía están estrechamente relacionados. Consideremos un gas ideal en equilibrio termodinámico. Un gas consta de una gran cantidad de moléculas que se mueven en un volumen V. Los parámetros del estado son la presión y la temperatura. El número de estados de tal sistema es enorme. La entropía de un gas en el equilibrio TD es máxima y, como se desprende de la fórmula de Boltzmann, está determinada por el número de microestados del sistema. Al mismo tiempo, no sabemos nada sobre qué estado específico tiene el sistema en un momento dado; la información es mínima. Digamos que de alguna manera logramos, usando un dispositivo muy rápido, “espiar el estado del sistema en un momento dado”. Entonces recibimos alguna información sobre ella. Incluso puedes imaginar que fotografiamos no solo las coordenadas de las moléculas, sino también sus velocidades (por ejemplo, tomando varias fotografías una tras otra). Además, en cada momento en el que disponemos de información sobre el estado del sistema, la entropía tiende a cero, porque el sistema sólo se encuentra en un estado específico entre toda su enorme variedad, y este estado es altamente no equilibrado. Este ejemplo muestra que la información y la entropía están efectivamente conectadas de alguna manera, y la naturaleza de la conexión ya está surgiendo: cuanta más información, menos entropía.
Información procedente de termodinámica y física estadística.
Las cantidades físicas que caracterizan los estados macroscópicos de los cuerpos (muchas moléculas) se denominan termodinámicas (incluida la energía, el volumen). Sin embargo, hay cantidades que aparecen como resultado de la acción de leyes puramente estadísticas y tienen significado cuando se aplican sólo a sistemas macroscópicos. Tales son, por ejemplo, la entropía y la temperatura.
Estadísticas clásicas
*Teorema de Liouville. La función de distribución es constante a lo largo de las trayectorias de fase del subsistema (estamos hablando de subsistemas cuasi cerrados, por lo que el teorema es válido solo para períodos de tiempo no muy grandes durante los cuales el subsistema se comporta como cerrado).

Aquí - - función de distribución o densidad de probabilidad. Se introduce a través de la probabilidad. w detectar un subsistema en un elemento del espacio de fase en este momento: dw = ( pag 1 ,..., PD , q 1 ,..., qs ) dpdq , y
Encontrar la distribución estadística de cualquier subsistema es la tarea principal de la estadística. Si se conoce la distribución estadística, entonces es posible calcular las probabilidades de diferentes valores de cualquier cantidad física dependiendo de los estados de este subsistema (es decir, de los valores de coordenadas y momentos):
![]() .
.
*Distribución microcanónica.
La distribución para el conjunto de dos subsistemas (se supone que son cerrados, es decir, que interactúan débilmente) es igual. Es por eso ![]() - logaritmo de la función de distribución - valor aditivo. Del teorema de Liouville se deduce que la función de distribución debe expresarse mediante combinaciones de variables p y q que, cuando el subsistema se mueve como un sistema cerrado, deben permanecer constantes (tales cantidades se denominan integrales de movimiento). Esto significa que la función de distribución en sí es una integral del movimiento. Además, su logaritmo también es una integral del movimiento, y aditivo. En total, en mecánica hay siete integrales del movimiento (energía, tres componentes del momento y tres componentes del momento angular) (para el subsistema a: mi (pag,
q),
PAG
A (pag,
q), METRO A (pag,
q)). La única combinación aditiva de estas cantidades es
- logaritmo de la función de distribución - valor aditivo. Del teorema de Liouville se deduce que la función de distribución debe expresarse mediante combinaciones de variables p y q que, cuando el subsistema se mueve como un sistema cerrado, deben permanecer constantes (tales cantidades se denominan integrales de movimiento). Esto significa que la función de distribución en sí es una integral del movimiento. Además, su logaritmo también es una integral del movimiento, y aditivo. En total, en mecánica hay siete integrales del movimiento (energía, tres componentes del momento y tres componentes del momento angular) (para el subsistema a: mi (pag,
q),
PAG
A (pag,
q), METRO A (pag,
q)). La única combinación aditiva de estas cantidades es
Además, los coeficientes (hay siete) deben seguir siendo los mismos para todos los subsistemas de un sistema cerrado determinado y se seleccionan a partir de las condiciones de normalización (4).
Para que se cumpla la condición de normalización (4), es necesario que la función (pag, q) contactado con los puntos mi 0, P 0, M 0 al infinito. Una formulación más precisa da la expresión
Distribución microcanónica.
La presencia de funciones - asegura que desaparezcan para todos los puntos del espacio de fase en el que al menos una de las cantidades MI, R,M no es igual a su valor dado (promedio) mi 0, P 0, M 0 .
De seis integrales PAG Y METRO puede eliminarse encerrando el sistema en una caja sólida en la que descanse.
![]() .
.
Entropía física
Nuevamente utilizamos el concepto de gas ideal.
Sea un gas ideal monoatómico con densidad. norte y temperatura t ocupa volumen V. Mediremos la temperatura en unidades de energía; la constante de Boltzmann no aparecerá. Cada átomo de gas tiene una energía cinética promedio de movimiento térmico igual a 3T/2. Por lo tanto, la energía térmica total del gas es igual a
Se sabe que la presión del gas es igual a pag = Nuevo Testamento. Si un gas puede intercambiar calor con el ambiente externo, entonces la ley de conservación de la energía del gas se ve así:
![]() . (5)
. (5)
Así, un cambio en la energía interna de un gas puede producirse tanto por el trabajo que realiza como por la recepción de una determinada cantidad de calor. dQ desde afuera. Esta ecuación expresa la primera ley de la termodinámica, es decir Ley de conservación de la energía. Se supone que el gas está en equilibrio, es decir pag = constante a lo largo de todo el volumen.
Si suponemos que el gas también está en un estado de equilibrio TD, t =constante, entonces la relación (5) puede considerarse como un proceso elemental de variación de los parámetros del gas cuando cambian muy lentamente, cuando el equilibrio TD no se altera. Es para tales procesos que se introduce el concepto de entropía S utilizando la relación
Así, se argumenta que, además de la energía interna, un gas en equilibrio tiene otra característica interna asociada con el movimiento térmico de los átomos. Según (5, 6) a volumen constante dV= 0, el cambio de energía es proporcional al cambio de temperatura, y en el caso general
![]()
Porque ![]() Dónde norte =
Nevada =
constante es el número total de átomos de gas, entonces la última relación se puede escribir en la forma
Dónde norte =
Nevada =
constante es el número total de átomos de gas, entonces la última relación se puede escribir en la forma
![]()
Después de la integración obtenemos

La expresión entre corchetes representa la entropía por partícula.
Por lo tanto, si tanto la temperatura como el volumen cambian de tal manera que Vermont 3/2 permanece constante, entonces la entropía S no cambia. Según (6), esto significa que el gas no intercambia calor con el ambiente externo, es decir el gas está separado de él por paredes aislantes del calor. Este proceso se llama adiabático.
Desde
donde = 5/3 se llama exponente adiabático. Por tanto, durante un proceso adiabático, la temperatura y la presión cambian con la densidad según la ley
fórmula de Boltzmann
Como se desprende del teorema de Liouville, ¿la función de distribución? tiene un máximo pronunciado en E = E 0 (valor promedio) y es distinto de cero solo en las proximidades de este punto. Si se ingresa el ancho E de la curva (E), definiéndolo como el ancho de un rectángulo cuya altura es igual al valor de la función (E) en el punto máximo, y el área es igual a la unidad ![]() (con la normalización adecuada). Podemos pasar del intervalo de valores de energía al número de estados Г con energías pertenecientes a E (esta es, de hecho, la fluctuación promedio de la energía del sistema). Entonces el valor Γ caracteriza el grado de mancha del estado macroscópico del sistema sobre sus estados microscópicos. En otras palabras, para los sistemas clásicos Г es el tamaño de la región del espacio de fases en la que un subsistema dado pasa casi todo su tiempo. En la teoría cuasi clásica, se establece una correspondencia entre el volumen de la región del espacio de fases y. el número de estados cuánticos por él. Es decir, para cada estado cuántico en el espacio de fases hay una celda con volumen , donde s es el número de grados de libertad.
(con la normalización adecuada). Podemos pasar del intervalo de valores de energía al número de estados Г con energías pertenecientes a E (esta es, de hecho, la fluctuación promedio de la energía del sistema). Entonces el valor Γ caracteriza el grado de mancha del estado macroscópico del sistema sobre sus estados microscópicos. En otras palabras, para los sistemas clásicos Г es el tamaño de la región del espacio de fases en la que un subsistema dado pasa casi todo su tiempo. En la teoría cuasi clásica, se establece una correspondencia entre el volumen de la región del espacio de fases y. el número de estados cuánticos por él. Es decir, para cada estado cuántico en el espacio de fases hay una celda con volumen , donde s es el número de grados de libertad.
El valor Γ se denomina peso estadístico del estado macroscópico y se puede escribir como:
El logaritmo del peso estadístico se llama entropía:
donde - peso estadístico = número de microestados cubiertos por el macroestado del sistema considerado.
![]() .
.
En estadística cuántica se muestra que = 1. Entonces
Donde por se entiende una matriz estadística (densidad). Debido a la linealidad del logaritmo de la función de distribución de energía (*), donde se realiza el promedio sobre la función de distribución.
Dado que el número de estados en cualquier caso no es menor que uno, la entropía no puede ser negativa. S determina la densidad de niveles en el espectro de energía de un sistema macroscópico. Debido a la aditividad de la entropía, podemos decir que las distancias promedio entre los niveles de un cuerpo macroscópico disminuyen exponencialmente con un aumento en su tamaño (es decir, el número de partículas en él). El valor de entropía más alto corresponde al equilibrio estadístico completo.
Al caracterizar cada estado macroscópico del sistema por la distribución de energía entre varios subsistemas, podemos decir que una serie de estados atravesados sucesivamente por el sistema corresponden a una distribución de energía cada vez más probable. Este aumento de probabilidad es grande debido a su naturaleza exponencial. ES- el exponente contiene una cantidad aditiva: entropía. Eso. Los procesos que ocurren en un sistema cerrado en desequilibrio proceden de tal manera que el sistema pasa continuamente de estados con menor entropía a estados con mayor entropía. Como resultado, la entropía alcanza el valor más alto posible, correspondiente al equilibrio estadístico completo.
Por lo tanto, si un sistema cerrado en algún momento se encuentra en un estado macroscópico de desequilibrio, entonces la consecuencia más probable en momentos posteriores será un aumento monótono en la entropía del sistema. Este - segunda ley de la termodinámica (R. Clausius, 1865). Su justificación estadística la dio L. Boltzmann en 1870. Otra definición:
Si en algún momento la entropía de un sistema cerrado difiere del máximo, entonces en los momentos siguientes la entropía no disminuye. Aumenta o, en el caso extremo, permanece constante. Según estas dos posibilidades, todos los procesos que ocurren con los cuerpos macroscópicos generalmente se dividen en irreversible Y reversible . Irreversible - aquellos procesos que van acompañados de un aumento en la entropía de todo el sistema cerrado (los procesos que se repetirían en orden inverso no pueden ocurrir, ya que en este caso la entropía tendría que disminuir). Tenga en cuenta que una disminución de la entropía puede deberse a fluctuaciones. Reversible Son procesos en los que la entropía de un sistema cerrado permanece constante y que, por tanto, también pueden ocurrir en sentido contrario. Un proceso estrictamente reversible representa un caso límite ideal.
Durante los procesos adiabáticos, el sistema no absorbe ni libera calor. ? q = 0 .
Comentario: (sustancial). La afirmación de que un sistema cerrado debe pasar a un estado de equilibrio en un tiempo suficientemente largo (más largo que el tiempo de relajación) se aplica sólo a un sistema en condiciones externas estacionarias. Un ejemplo es el comportamiento de una gran región del Universo accesible a nuestra observación (las propiedades de la naturaleza no tienen nada en común con las propiedades de un sistema en equilibrio).
Información.
Consideremos una cinta dividida en celdas: un registro clásico. Si solo se puede colocar uno de dos caracteres en cada celda, entonces se dice que la celda contiene un poco de información. Es obvio (ver lección 1) que en el registro que contiene norte células contenidas norte Un poco de información y se puede escribir en él. 2 norte mensajes. Entonces, la entropía de la información se mide en bits:
![]() (7)
(7)
Aquí QN = 2 norte- el número total de mensajes diferentes. De (7) está claro que la entropía de la información es simplemente igual al número mínimo de celdas binarias con las que se puede registrar cierta información.
La definición (7) se puede reescribir de otra manera. tengamos muchos QN varios mensajes. Encontremos la probabilidad de que el mensaje que necesitamos coincida con uno seleccionado al azar del número total. QN varios mensajes. obviamente es igual a PN = 1/ QN. Entonces la definición (7) se escribirá como:
![]() (8)
(8)
Cuanto mayor sea el número de células norte, menos probable es PN y cuanto mayor sea la entropía de la información HB contenido en este mensaje en particular.
Ejemplo . El número de letras del alfabeto es 32 (sin la letra ё). El número 32 es la quinta potencia de dos 32 = 2 5. Para hacer coincidir cada letra con una combinación específica de números binarios, necesita tener 5 celdas. Al agregar letras mayúsculas a minúsculas, duplicamos la cantidad de caracteres que queremos codificar; habrá 64 = 2 6, es decir, se agrega un poco más de información HB= 6. Aquí HB- la cantidad de información por letra (minúscula o mayúscula). Sin embargo, un cálculo tan directo de la entropía de la información no es del todo exacto, ya que hay letras en el alfabeto que son más o menos comunes. A las letras que aparecen con menos frecuencia se les puede asignar una mayor cantidad de celdas, y para las letras que ocurren con frecuencia, se puede ahorrar dinero y darles esos estados de registro que ocupan una menor cantidad de celdas. Shannon dio la definición exacta de entropía de la información:
![]() (9)
(9)
Formalmente, la derivación de esta relación se puede justificar de la siguiente manera.
Mostramos arriba eso
debido a la aditividad del logaritmo de la función de distribución y su linealidad en energía.
Dejar pag- función de distribución de algún valor discreto f i (por ejemplo, la letra "o" en este texto). Si usa la función pag construir la función de distribución de probabilidad de varios valores de la cantidad F = F 1 , F 2 ,... f norte, entonces esta función tendrá un máximo en , donde y (normalización). Entonces p()= 1 y (en términos generales, esto es cierto para la clase de funciones que satisfacen la condición (*))
La suma se realiza sobre todos los caracteres (letras del alfabeto), y p yo significa la probabilidad de que aparezca un símbolo con un número i. Como puede ver, esta expresión abarca tanto letras de uso frecuente como letras cuya probabilidad de aparecer en un mensaje determinado es baja.
Dado que la expresión (9) utiliza el logaritmo natural, la unidad de información correspondiente se denomina “nat”.
La expresión (9) se puede reescribir como
donde los paréntesis significan el promedio clásico habitual utilizando la función de distribución p i.
Comentario . En las siguientes conferencias se demostrará que para los estados cuánticos
¿Dónde está la matriz de densidad? Formalmente, las expresiones (10) y (11) son iguales, pero hay una diferencia significativa. El promedio clásico se realiza sobre estados ortogonales (eigen) del sistema, mientras que para el caso cuántico también puede haber estados no ortogonales (superposiciones). Por eso siempre H cuanto clase H !
Las fórmulas (8) y (9) utilizan logaritmos en diferentes bases. En (8), basado en la base 2, y en (9), basado en la base e, las entropías de información correspondientes a estas fórmulas se pueden expresar fácilmente entre sí. Usemos la relación en la que M es un número arbitrario.
![]() .
.
Entonces, considerando que ![]() y obtenemos
y obtenemos
![]() - ¡La cantidad de bits es casi una vez y media mayor que la cantidad de nat!
- ¡La cantidad de bits es casi una vez y media mayor que la cantidad de nat!
Razonando de manera similar, podemos obtener la relación entre entropías expresadas en trits y bits:
En tecnología informática, la información se utiliza en base binaria (en bits). Para el razonamiento en física, es más conveniente utilizar la información de Shannon (en Nat), que puede caracterizar cualquier información discreta. Siempre puedes encontrar el número de bits correspondientes.
RELACIÓN DE ENTROPÍA E INFORMACIÓN. El demonio de Maxwell
Esta paradoja fue considerada por primera vez por Maxwell en 1871 (ver Fig. 1). Dejar que una fuerza “sobrenatural” abra y cierre la válvula en un recipiente dividido en dos partes y que contiene gas. La válvula está controlada por la regla de que se abre si la tocan moléculas rápidas que se mueven de derecha a izquierda o si la golpean moléculas lentas que se mueven en la dirección opuesta. Así, el demonio introduce una diferencia de temperatura entre dos volúmenes sin realizar trabajo, lo que viola la segunda ley de la termodinámica.

El demonio de Maxwell. El demonio establece una diferencia de presión abriendo la compuerta cuando el número de moléculas de gas que lo golpean desde la izquierda excede el número de golpes desde la derecha. Esto se puede hacer de una manera completamente reversible, siempre y cuando los resultados aleatorios del demonio de sus observaciones de moléculas estén almacenados en su memoria. Por tanto, la memoria del demonio (o su cabeza) se calienta. El paso irreversible no es que la información se acumule, sino que la información se pierda cuando el demonio luego borra la memoria. Arriba: Llenar la memoria del demonio con bits de información es un proceso aleatorio. En el lado derecho de la línea de puntos hay un área de memoria vacía (todas las celdas están en el estado 0, a la izquierda hay bits aleatorios). A continuación se muestra un demonio.
Se han hecho varios intentos para resolver la paradoja o exorcizar al demonio. Por ejemplo, se suponía que el demonio no podía extraer información sin trabajar o sin perturbar (es decir, calentar) el gas, ¡pero resultó que no era así! Otros intentos se redujeron al hecho de que el segundo principio podría violarse bajo la influencia de ciertas fuerzas (criaturas) "razonables" o "pensantes". En 1929 Leo Szilard “avanzó” significativamente la solución al problema, reduciéndola a una formulación mínima y destacando los componentes esenciales. Lo principal que debe hacer el Demonio es establecer si a la derecha o a la izquierda de la válvula deslizante hay una sola molécula que permitiría extraer el calor. Este dispositivo se llamó motor Szilard. Sin embargo, Szilard no resolvió la paradoja porque su análisis no tuvo en cuenta cómo la medida por la que el demonio sabe si una molécula está a la derecha o a la izquierda afecta al aumento de la entropía (ver figura Szilard_demon.pdf). El motor funciona en un ciclo de seis pasos. El motor es un cilindro con pistones en los extremos. Se inserta una solapa en el medio. El trabajo de mover la partición se puede reducir a cero (Szilard lo demostró). También hay un dispositivo de memoria (MU). Puede estar en uno de tres estados. "Vacío", "Molécula de la derecha" y "Molécula de la izquierda". Estado inicial: ARRIBA = “Vacío”, los pistones están presionados hacia afuera, la partición está extendida, la molécula tiene una velocidad promedio, que está determinada por la temperatura del termostato (diapositiva 1).
1. Se inserta el tabique dejando la molécula a la derecha o a la izquierda (diapositiva 2).
2. El dispositivo de memoria determina dónde está la molécula y pasa al estado "derecha" o "izquierda".
3. Compresión. Dependiendo del estado del UE, el pistón entra desde el lado donde no hay molécula. Esta etapa no requiere ningún trabajo por realizar. Porque el vacío se comprime (diapositiva 3).
4. Se retira el tabique. La molécula comienza a ejercer presión sobre el pistón (diapositiva 4).
5. Carrera de trabajo. La molécula golpea el pistón y hace que se mueva en la dirección opuesta. La energía de la molécula se transfiere al pistón. A medida que el pistón se mueve, su velocidad promedio debería disminuir. Sin embargo, esto no sucede, ya que las paredes del recipiente están a temperatura constante. Por tanto, el calor del termostato se transfiere a la molécula, manteniendo constante su velocidad. Así, durante la carrera de trabajo, la energía térmica suministrada por el termostato se convierte en trabajo mecánico realizado por el pistón (corredera 6).
6. Limpiar el UE y devolverlo al estado "Vacío" (diapositiva 7). El ciclo se completa (diapositiva 8 = diapositiva 1).
Es sorprendente que esta paradoja no se resolvió hasta los años 1980. Durante este tiempo se estableció que, en principio, cualquier proceso puede realizarse de forma reversible, es decir. sin “pago” por entropía. Finalmente, Bennett en 1982 estableció la conexión definitiva entre esta afirmación y la paradoja de Maxwell. Propuso que el demonio podía saber realmente dónde estaba una molécula en el motor de Szilard sin realizar trabajo ni aumentar la entropía del entorno (el termostato) y, por tanto, realizar un trabajo útil en un ciclo del motor. Sin embargo, la información sobre la posición de la molécula debe permanecer en la memoria del demonio (rsi.1). A medida que se realizan más ciclos, se acumula más y más información en la memoria. Para completar el ciclo termodinámico, el demonio debe borrar la información almacenada en la memoria. Es esta operación de borrar información la que debe clasificarse como un proceso de aumento de la entropía del medio ambiente, como exige el segundo principio. Esto completa la parte fundamentalmente física del dispositivo del demonio de Maxwell.
Estas ideas recibieron cierto desarrollo en las obras de D.D. Kadomtsev.
Consideremos un gas ideal formado por una sola partícula (Kadomtsev, “dinámica e información”). Esto no es absurdo. Si una partícula está encerrada en un recipiente de volumen V con paredes a temperatura T, tarde o temprano entrará en equilibrio con estas paredes. En cada momento del tiempo está en un punto muy concreto del espacio y a una velocidad muy concreta. Llevaremos a cabo todos los procesos tan lentamente que la partícula tendrá tiempo, en promedio, para llenar todo el volumen y cambiar repetidamente la magnitud y dirección de la velocidad durante colisiones inelásticas con las paredes del recipiente. Así, la partícula ejerce una presión promedio sobre las paredes y tiene una temperatura t y su distribución de velocidades es Maxwelliana con temperatura t. Este sistema de una partícula se puede comprimir adiabáticamente, se puede cambiar su temperatura, dándole la oportunidad de alcanzar el equilibrio con las paredes del recipiente.
Presión promedio sobre la pared en norte = 1 , es igual pag=T/V, y la densidad promedio n = 1/ V. Consideremos el caso de un proceso isotérmico cuando t =constante. Desde el primer comienzo en t =constante. Y pag=T/V obtenemos
![]() , porque
, porque
De aquí encontramos que el cambio de entropía no depende de la temperatura, por lo que
![]()
Aquí se introduce la constante de integración: “tamaño de partícula”< Trabajar en un proceso isotérmico. El trabajo está determinado por la diferencia de entropía. Supongamos que tenemos particiones ideales que se pueden usar para dividir el recipiente en partes sin desperdiciar energía. Dividamos nuestro recipiente en dos partes iguales con un volumen. V/2
cada. En este caso, la partícula estará en una de las mitades, pero no sabemos cuál. Digamos que tenemos un dispositivo que nos permite determinar en qué parte se encuentra una partícula, por ejemplo, una balanza de precisión. Luego, a partir de una distribución de probabilidad simétrica del 50% al 50% dividida en dos mitades, obtenemos una probabilidad del 100% para una de las mitades; se produce un "colapso" de la distribución de probabilidad. En consecuencia, la nueva entropía será menor que la entropía original en la cantidad Al disminuir la entropía, se puede realizar trabajo. Para ello basta con mover la partición hacia el volumen vacío hasta que desaparezca. El trabajo será igual a Si nada ha cambiado en el mundo exterior, entonces, repitiendo estos ciclos, es posible construir una máquina de movimiento perpetuo del segundo tipo. Este es el demonio de Maxwell en la versión de Szilard. Pero la segunda ley de la termodinámica prohíbe obtener trabajo únicamente a través del calor. Esto significa que algo debe estar sucediendo en el mundo exterior. ¿Qué es esto? Detección de una partícula en una de las mitades. cambia la información sobre una partícula -
De las dos posibles mitades, sólo se indica una, en la que se encuentra la partícula. Este conocimiento corresponde a un bit de información. El proceso de medición reduce la entropía de la partícula (transferencia a un estado de desequilibrio) y aumenta la información sobre el sistema (partícula) exactamente en la misma cantidad. Si divide repetidamente por la mitad las mitades, cuartos, octavos, etc. obtenidos anteriormente, la entropía disminuirá constantemente y la información aumentará. En otras palabras Cuanto más se sabe sobre un sistema físico, menor es su entropía. Si se sabe todo sobre el sistema, esto significa que lo hemos trasladado a un estado de alto desequilibrio, cuando sus parámetros están lo más lejos posible de los valores de equilibrio. Si en nuestro modelo la partícula se puede colocar en una celda elemental de volumen V 0
, entonces al mismo tiempo S = 0
, y la información alcanza su valor máximo Entonces, la entropía de la información. es una medida de la falta (o grado de incertidumbre) de información sobre el estado real de un sistema físico. Entropía de información de Shannon: cantidad de información
I(o simplemente información) sobre el estado de un sistema clásico, obtenida como resultado de mediciones realizadas por un dispositivo externo conectado al sistema considerado por algún canal de comunicación, se define como la diferencia en la entropía de la información correspondiente a la incertidumbre inicial del sistema. estado h 0
, y entropía de información del estado final del sistema después de la medición. h. De este modo, I
+
h
=
h
0
=
constante
.
En el caso ideal, cuando no hay ruido ni interferencias creadas por fuentes externas en el canal de comunicación, la distribución de probabilidad final después de la medición se reduce a un valor específico. pn= 1, es decir h =
0, y se determinará el valor máximo de la información obtenida durante la medición:
imax =
h 0
. Así, la entropía de información de Shannon de un sistema tiene el significado de la información máxima contenida en el sistema; se puede determinar en condiciones ideales de medición del estado del sistema en ausencia de ruido e interferencias, cuando la entropía del estado final es cero: Consideremos un elemento lógico clásico que puede estar en uno de dos estados lógicos igualmente probables "0" y "1". Dicho elemento, junto con el entorno (el termostato y la señal generada por un objeto externo aislado térmicamente), forma un único sistema cerrado en desequilibrio. La transición de un elemento a uno de los estados, por ejemplo, al estado "0", corresponde a una disminución de las estadísticas. el peso de su estado en comparación con el estado inicial es 2 veces (para sistemas de tres niveles, 3 veces). Encontremos la disminución entropía de la información Shannon, que corresponde a un aumento en la cantidad de información sobre un elemento en uno, lo que se llama poco: Por tanto, la entropía de la información determina la cantidad de bits que se requieren para codificar la información en el sistema o mensaje en cuestión. LITERATURA 1. D. Landau, I. Lifshits. Física estadística. Parte 1. Ciencia, M 1976. 2. MA Leontovich. Introducción a la termodinámica. Física estadística. Moscú, Nauka, 1983. - 416 p. 3. B. B. Kadomtsev. Dinámica e información. UFN, 164, núm. 5, 449 (1994). TERMODINÁMICA ESTADÍSTICA, sección estadística. física, dedicada a la fundamentación de las leyes de la termodinámica a partir de las leyes de interacción. y los movimientos de las partículas que componen el sistema. Para sistemas en estado de equilibrio, la termodinámica estadística permite calcular potenciales termodinámicos, escribir ecuaciones de estado, fase y condiciones químicas. equilibrios. La termodinámica estadística de desequilibrio proporciona justificación de las relaciones (ecuaciones de transferencia de energía, momento, masa y sus condiciones de contorno) y permite calcular la cinética incluida en las ecuaciones de transferencia. coeficientes La termodinámica estadística establece cantidades. conexión entre micro y macropropiedades físicas. y química. sistemas Los métodos de cálculo de la termodinámica estadística se utilizan en todas las áreas de la ciencia moderna. teorético química microcanónico El conjunto de Gibbs se utiliza cuando se consideran sistemas aislados (que no intercambian energía E con el medio ambiente), que tienen un volumen constante V y un número de partículas idénticas N (E, V y N son los parámetros del estado del sistema). Kanonich. El conjunto de Gibbs se utiliza para describir sistemas de volumen constante que están en equilibrio térmico con el medio ambiente (temperatura absoluta T) con un número constante de partículas N (parámetros de estado V, T, N). Gran Canon. El conjunto de Gibbs se utiliza para describir sistemas abiertos que están en equilibrio térmico con el medio ambiente (temperatura T) y equilibrio material con un depósito de partículas (las partículas de todo tipo se intercambian a través de las “paredes” que rodean el sistema con volumen V). Los parámetros de estado de dicho sistema son V, T y m: el potencial químico de las partículas. isobárico-isotermal El conjunto de Gibbs se utiliza para describir sistemas térmicos y de piel. equilibrio con el medio ambiente a presión constante P (parámetros de estado T, P, N). Espacio de fase en estadística. La mecánica es un espacio multidimensional, cuyos ejes son todas las coordenadas generalizadas q i y los impulsos asociados p i (i = 1,2,..., M) de un sistema con M grados de libertad. Para un sistema que consta de N átomos, q i y p i corresponden a la coordenada cartesiana y al componente de momento (a = x, y, z) de un determinado átomo j y M = 3N. El conjunto de coordenadas y momentos se denotan por q y p, respectivamente. El estado del sistema está representado por un punto en un espacio de fase de dimensión 2M, y el cambio en el estado del sistema en el tiempo está representado por el movimiento de un punto a lo largo de una línea, llamada. trayectoria de fase. Para estadística Para describir el estado del sistema, se introducen los conceptos de volumen de fase (un elemento del volumen del espacio de fase) y la función de distribución f(p, q), que caracteriza la densidad de probabilidad de encontrar un punto que represente el estado del sistema. sistema en un elemento del espacio de fase cerca de un punto con coordenadas p, q. En mecánica cuántica, en lugar del volumen de fase, se utiliza el concepto de energía discreta. espectro de un sistema de volumen finito, porque el estado de una partícula individual no está determinado por el momento y las coordenadas, sino por una función de onda, un corte en la dinámica estacionaria. el estado del sistema corresponde a la energía. espectro de estados cuánticos. Función de distribuciónclásico El sistema f(p, q) caracteriza la densidad de probabilidad de la implementación de un micro dado. donde dГ N es el elemento del volumen de fase del sistema en unidades de h 3N, h es la constante de Planck; divisor norte! tiene en cuenta el hecho de que la reordenación de las identidades. Las partículas no cambian el estado del sistema. La función de distribución satisface la condición de normalización t f(p, q)dГ N = 1, porque el sistema es confiable en el k.-l. condición. Para los sistemas cuánticos, la función de distribución determina la probabilidad w i, N de encontrar un sistema de N partículas en un estado cuántico especificado por un conjunto de números cuánticos i, con energía E i, N, sujeto a normalización. El valor medio en el momento t (es decir, segúnintervalo de tiempo infinitamente pequeño de t a t + dt) cualquier físico. el valor A(p, q), que es función de las coordenadas y momentos de todas las partículas del sistema, se calcula utilizando la función de distribución según la regla (incluso para procesos de desequilibrio): La integración sobre coordenadas se realiza en todo el volumen del sistema y la integración sobre impulsos desde - , hasta +, . Estado termodinámico el equilibrio del sistema debe considerarse como el límite t: , . Para estados de equilibrio, las funciones de distribución se determinan sin resolver la ecuación de movimiento de las partículas que forman el sistema. La forma de estas funciones (la misma para los sistemas clásicos y cuánticos) fue establecida por J. Gibbs (1901). En microcanon. En el conjunto de Gibbs, todos los microestados con una energía dada E son igualmente probables y la función de distribución para el clásico sistemas tiene la forma: f(p,q) = A d, Dónde d - Función delta de Dirac, H(p, q) - Función de Hamilton, que es la suma de la cinética. y potencial energías de todas las partículas; la constante A se determina a partir de la condición de normalización de la función f(p, q). Para sistemas cuánticos, con la precisión de especificar el estado cuántico igual al valor D E, de acuerdo con la relación de incertidumbre entre energía y tiempo (entre el momento y las coordenadas de las partículas), función w (E k) = -1, si EE k E + D E, y w (E k) = 0 si E k< Е и E k >E + D E. Valor g(E, N, V)-t. llamado estadístico peso igual al número de estados cuánticos en la energía. capa D E. Una relación importante de la termodinámica estadística es la conexión entre la entropía del sistema y la estadística. peso: S(E, N, V) = klng(E, N, V), donde la constante k-Boltzmann. En canon. En el conjunto de Gibbs, la probabilidad de que el sistema esté en un microestado determinado por las coordenadas y momentos de todas las N partículas o los valores de E i,N tiene la forma: f(p, q) = exp (/kT) ;w i,N = exp[(F - E i,N)/kT], donde F-libre. dónde Z N -estadístico. suma (en el caso de un sistema cuántico) o estadística. integral (en el caso de un sistema clásico), determinada a partir de la condición de normalización de las funciones w i,N o f(p, q): Z norte = t exp[-H(p, q)/kT]dpdq/(N!h 3N) (la suma de r se toma en todos los estados cuánticos del sistema y la integración se lleva a cabo en todo el espacio de fases). Dónde En el gran canon. Función de distribución del conjunto de Gibbs f(p, q) y estadística. la suma X, determinada a partir de la condición de normalización, tiene la forma: W - termodinámico Para calcular la termodinámica potencial dependiendo de las variables V, T, m (la suma se realiza sobre todos los números enteros positivos N). En isobárico-isotermo Distribución del conjunto de Gibbs y función estadística. la suma Q, determinada a partir de la condición de normalización, tiene la forma: donde G es la energía de Gibbs del sistema (potencial isobárico-isotérmico, entalpía libre).Sistemas ideales. Cálculo de estadísticas. Las sumas de la mayoría de los sistemas es una tarea difícil. Se simplifica notablemente en el caso de los gases, si se trata del aporte de potencial. la energía en la energía total del sistema puede despreciarse. En este caso, la función de distribución completa f(p, q) para N partículas de un sistema ideal se expresa mediante el producto de las funciones de distribución de una sola partícula f 1 (p, q): La distribución de partículas entre microestados depende de su cinética. energía y de los santos cuánticos en el sistema, debido a debido a la identidad de las partículas. En mecánica cuántica, todas las partículas se dividen en dos clases: fermiones y bosones. El tipo de estadística que obedecen las partículas está únicamente relacionado con su espín. La estadística de Fermi-Dirac describe la distribución en un sistema de identidades. partículas con espín semientero 1/2, 3/2,... en unidades ђ = h/2p. Se llama una partícula (o cuasipartícula) que obedece a las estadísticas especificadas. fermión. Los fermiones incluyen electrones en átomos, metales y semiconductores, núcleos atómicos con un número atómico impar, átomos con una diferencia impar entre el número atómico y el número de electrones, cuasipartículas (por ejemplo, electrones y huecos en sólidos), etc. Esta estadística fue propuesta por E. Fermi en 1926; Ese mismo año, P. Dirac descubrió su mecánica cuántica. significado. La función de onda del sistema de fermiones es antisimétrica, es decir cambia de signo al reorganizar las coordenadas y giros de cualquier par de identidades. partículas. No puede haber más de una partícula en cada estado cuántico (ver principio de Pauli). Las estadísticas de Bose-Einstein describen sistemas de identidades. partículas con espín cero o entero (0, ђ, 2ђ, ...). Se llama partícula o cuasipartícula que obedece a las estadísticas especificadas. bosón. Esta estadística fue propuesta por S. Bose (1924) para los fotones y desarrollada por A. Einstein (1924) en relación con las moléculas de gases ideales, consideradas como partículas compuestas de un número par de fermiones, por ejemplo. núcleos atómicos con un número total par de protones y neutrones (deuterón, núcleo de 4 He, etc.). Los bosones también incluyen fonones en 4 He sólido y líquido, excitones en semiconductores y dieléctricos. La función de onda del sistema es simétrica con respecto a la permutación de cualquier par de identidades. partículas. Los números de ocupación de los estados cuánticos no están limitados por nada, es decir. Cualquier número de partículas puede existir en un estado. El número promedio de partículas n i de un gas ideal de bosones en un estado con energía E i se describe mediante la función de distribución de Bose-Einstein: n yo =(exp[(E yo - m )/kT]-1) -1 . La estadística de Boltzmann es un caso especial de la estadística cuántica, cuando los efectos cuánticos pueden despreciarse (altas temperaturas). Considera la distribución de las partículas de gas ideal en momento y coordenadas en el espacio de fases de una partícula, y no en el espacio de fases de todas las partículas, como en las distribuciones de Gibbs. Como minimo Unidades de volumen del espacio de fases, que tiene seis dimensiones (tres coordenadas y tres proyecciones del momento de las partículas), de acuerdo con la mecánica cuántica. debido a la relación de incertidumbre, es imposible elegir un volumen menor que h 3 . El número promedio de partículas n i de un gas ideal en un estado con energía E i se describe mediante la función de distribución de Boltzmann:n yo =exp[( m -E i)/kT]. Para partículas que se mueven según leyes clásicas. mecanica en exterior potencial En el campo U(r), la función estadística de equilibrio de la distribución f 1 (p,r) sobre los momentos p y las coordenadas r de las partículas de gas ideal tiene la forma: f 1 (p,r) = A exp( - [p 2 /2m + U(r)]/kT).se enfría en un campo gravitacional (f-la barométrico), moléculas y partículas altamente dispersas en un campo de fuerzas centrífugas, electrones en semiconductores no degenerados, y también se utiliza para calcular la distribución de iones en un estado diluido. soluciones de electrolitos (a granel y en el límite con el electrodo), etc. Con U(r) = 0, la distribución de Maxwell-Boltzmann se deriva de la distribución de Maxwell-Boltzmann, que describe la distribución de velocidades de las partículas en un formato estadístico. estado. equilibrio (J. Maxwell, 1859). Según esta distribución, el número probable de moléculas por unidad de volumen, cuyas componentes de velocidad se encuentran en los intervalos de u i a u i + du i (i = x, y, z), está determinado por la siguiente función: La distribución de Maxwell no depende de la interacción. entre partículas y es válido no sólo para los gases, sino también para los líquidos (si es posible una descripción clásica para ellos), así como para las partículas brownianas suspendidas en líquidos y gases. Se utiliza para contar el número de colisiones de moléculas de gas entre sí durante reacciones químicas. r-ción y con átomos de superficie. Suma de estados de una molécula. Estadístico suma de un gas ideal en canónico El conjunto de Gibbs se expresa mediante la suma de los estados de una molécula Q 1: donde E i es la energía del i-ésimo nivel cuántico de la molécula (i = O corresponde al nivel cero de la molécula), g i es estadístico. peso del i-ésimo nivel. Dónde En el caso general, los tipos individuales de movimiento de electrones, átomos y grupos de átomos en una molécula, así como el movimiento de la molécula en su conjunto, están interrelacionados, pero aproximadamente pueden considerarse independientes. Entonces la suma de los estados de la molécula podría ser presentado en forma de un producto de componentes individuales asociados con los pasos. movimiento (Q post) y con intramol. movimientos (Q int): La suma de los estados de movimiento electrónico Q el es igual a estadística. peso P t base. Estado electrónico de una molécula. En plural casos bas. el nivel no es degenerado y está separado del nivel excitado más cercano, es decir. energía: (P t = 1).Sin embargo, en algunos casos, p. para la molécula de O 2, Р t = з, básicamente. estado, el momento de movimiento de la molécula es diferente de cero y hay degeneración de los niveles de energía, y las energías de los estados excitados pueden hacerlo. bastante bajo. La suma de los estados del veneno Q, debido a la degeneración de los espines nucleares, es igual a:donde s i es el espín del núcleo del átomo i, el producto se toma entre todos los átomos de la molécula. Suma por estados de oscilación. movimiento pequeñas fluctuaciones, n es el número de átomos en una molécula. La suma por estado será rotada. Los movimientos de una molécula poliatómica con grandes momentos de inercia se pueden considerar clásicamente [aproximación de alta temperatura, T/q i 1, donde q i = h 2 /8p 2 kI i (i = x, y, z), I t es el principal momento de inercia de rotación alrededor del eje i] : Q tiempo = (p T 3 /q x q y q z) 1/2.Para moléculas lineales con un momento de inercia I estadístico. suma Q tiempo = T/q, donde q = h 2 /8p 2 *kI. Al calcular a temperaturas superiores a 10 3 K, es necesario tener en cuenta la anarmonicidad de las vibraciones atómicas y los efectos de interacción. oscilar y girar. grados de libertad (ver Moléculas no rígidas), así como multiplicidad de estados electrónicos, población de niveles excitados, etc. A bajas temperaturas (por debajo de 10 K), es necesario tener en cuenta los efectos cuánticos (especialmente para moléculas diatómicas) . Bien, vamos a rotarlo. El movimiento de una molécula AB heteronuclear se describe mediante la siguiente fórmula: rotación del número l. estados, y para moléculas homonucleares A 2 (especialmente para moléculas de hidrógeno H 2, deuterio D 2, tritio T 2) nucleares y rotativas. grados de libertad de interacción amigo En los gases reales, las moléculas interactúan. unos con otros. En este caso, la suma de los estados del conjunto no se reduce al producto de las sumas de los estados de las moléculas individuales. Si asumimos que intermol. interacción no afecta interna estados de moléculas, estadísticos suma del sistema en clásico aproximación para un gas que consta de N identidades. partículas tiene la forma: Dónde Aquí<2 N-config.integral teniendo en cuenta la interacción. moléculas Naib, a menudo potencial. la energía de las moléculas U se considera como la suma de potenciales de pares: U = = donde U(r ij) es el potencial central. fuerzas dependiendo de distancias r ij entre las moléculas i y j. También se tienen en cuenta las contribuciones de múltiples partículas al potencial. energía, efectos de orientación molecular, etc. La necesidad de calcular la configuración. integral surge al considerar cualquier condensador. fases y límites de fase. Solución exacta al problema del plural. órganos es casi imposible, por lo tanto, calcular datos estadísticos. sumas y todo termodinámico. St. in, obtenido de estadística. sumas por diferenciación según los parámetros correspondientes, utilice diff. métodos aproximados. Según el llamado Método de expansión de grupos, el estado del sistema se considera como un conjunto de complejos (grupos) que constan de un número diferente de moléculas y configuraciones. la integral se descompone en un conjunto de integrales de grupo. Este enfoque nos permite imaginar cualquier termodinámica. f-ción del gas real en forma de una serie de grados de densidad.dos partículas en los puntos 1 y 2, determina el llamado. función de correlación g(|r 1 - r 2 |) = f 2 (r 1, r 2)/r 2, que caracteriza la correlación mutua en la distribución de partículas. La información experimental correspondiente se obtiene mediante análisis estructural de rayos X. Las funciones de distribución de dimensiones n y n + 1 están conectadas por un sistema infinito de integrodiferenciales entrelazados. Ecuaciones de Bogolyubov-Born-Green-Kirkwood-Yvon, cuya solución es extremadamente difícil, por lo que se tienen en cuenta los efectos de la correlación entre partículas mediante la introducción de la descomposición. aproximaciones, que determinan cómo se expresa la función f n a través de funciones de menor dimensión. Resp. desarrollado por varios métodos aproximados para calcular funciones f n, y a través de ellos todos termodinámicos. características del sistema considerado. Naib. Se utilizan las aproximaciones de Perkus-Ievik y de hipercadena. Modelos de condensadores de celosía.Los estados han encontrado una amplia aplicación en termodinámica. consideración de casi todo lo físico-químico. tareas. Todo el volumen del sistema se divide en regiones locales con un tamaño característico del orden del tamaño de la molécula u 0 . En general, en diferentes modelos el tamaño del área local puede ser tanto mayor como menor que u 0 ;en la mayoría de los casos son iguales. La transición a una distribución discreta de moléculas en el espacio simplifica significativamente el cálculo de la descomposición. configuraciones moleculares. Los modelos de celosía tienen en cuenta la interacción. moléculas entre sí; interacción energética descrito energéticamente. parámetros. En varios casos, los modelos reticulares permiten soluciones exactas, lo que permite evaluar la naturaleza de las aproximaciones utilizadas. Con su ayuda, es posible considerar multipartículas y específicas. interacción, orientación efectos, etc. Los modelos de celosía son fundamentales en el estudio e implementación de cálculos aplicados de soluciones de polímeros y no electrolitos, transiciones de fase, fenómenos críticos y sistemas altamente heterogéneos. Métodos numéricos para determinar la termodinámica. St.-in se están volviendo cada vez más importantes a medida que se desarrolla la informática. En el método dicen. En dinámica, la evolución del estado del sistema se considera utilizando la integración numérica de las ecuaciones de Newton para el movimiento de cada partícula (N = = 10 2 -10 5) a potenciales dados de interacción entre partículas. Las características de equilibrio del sistema se obtienen promediando trayectorias de fase (velocidades y coordenadas) durante largos tiempos, después de establecer la distribución maxwelliana de partículas sobre velocidades (el llamado período de termalización). Limitaciones en el uso de métodos numéricos en básico. determinado por las capacidades de la computadora. Especialista. calculará. las técnicas le permiten evitar las dificultades asociadas con el hecho de que no se está considerando un sistema real, sino un pequeño volumen; Esto es especialmente importante cuando se tienen en cuenta los potenciales de interacción de largo alcance, se analizan las transiciones de fase, etc. La cinética física es una sección de la estadística. física, que proporciona una justificación de las relaciones de la termodinámica de procesos irreversibles, que describen la transferencia de energía, momento y masa, así como la influencia de influencias externas en estos procesos. campos. Cinético. coeficientes macroscópicos Características de un medio continuo que determinan las dependencias de los flujos físicos. cantidades (calor, momento, masa de componentes, etc.) deprovocando estos flujos de gradientes, concentraciones, hidrodinámica. velocidad, etc. Es necesario distinguir los coeficientes de Onsager incluidos en las ecuaciones que conectan los flujos con la termodinámica. fuerzas (ecuación termodinámica de movimiento) y coeficientes de transferencia (difusión, conductividad térmica, viscosidad, etc.) incluidos en la ecuación de transferencia. El primer m.b. expresado a través de este último utilizando las relaciones entre macroscópicas. características del sistema, por lo que en el futuro sólo se considerarán coeficientes. transferir. Para ellos, tal vez. Se ha compilado un sistema de ecuaciones que permite describir estados arbitrarios de desequilibrio. Resolver este sistema de ecuaciones es muy difícil. Como regla general, en cinética. La teoría de los gases y cuasipartículas gaseosas en sólidos (fermiones y bosones) utiliza únicamente la ecuación para la función de distribución de una sola partícula f 1. Bajo el supuesto de que no existe correlación entre los estados de ninguna partícula (hipótesis del caos molecular), se crea el llamado cinético Ecuación de Boltzmann (L. Boltzmann, 1872). Esta ecuación tiene en cuenta el cambio en la distribución de partículas bajo la influencia de influencias externas. fuerzas F(r, m) y colisiones de pares entre partículas: Dónde f 1 (u, r, t) y funciones de distribución de partículas hastacolisiones, f " 1 (u", r, t) y funciones de distribucióndespués de una colisión; u y -velocidades de las partículas antes de la colisión, u" y -velocidades de las mismas partículas después de la colisión, y = |u -|-módulo de la velocidad relativa de las partículas en colisión, q - el ángulo entre la velocidad relativa de las u - partículas en colisión y la línea que conecta sus centros , s (u,q )dW - sección transversal diferencial de la dispersión de partículas por el ángulo sólido dW en el sistema de coordenadas de laboratorio, dependiendo de la ley de las partículas. Para el modelo de moléculas en la forma. de esferas rígidas elásticas con radio R, se supone s = 4R 2. En el marco de la mecánica clásica, la sección transversal diferencial se expresa en términos de los parámetros de colisión b y e (la distancia de impacto correspondiente y el ángulo acimutal de la esfera). línea de centros): s dW = bdbde, y las moléculas se consideran como centros de fuerzas con un potencial que depende de la distancia. La expresión para la sección transversal efectiva diferencial se obtiene con base en la mecánica cuántica, teniendo en cuenta la influencia de la simetría. efectos sobre la probabilidad de colisión. Si el sistema está en estado estadístico. En equilibrio, la integral de colisión Stf es igual a cero y la solución es cinética. La ecuación de Boltzmann será la distribución de Maxwell. Para estados de desequilibrio, las soluciones de la cinética. Las ecuaciones de Boltzmann generalmente se buscan en forma de una expansión en serie de la función f 1 (u, r, m) en parámetros pequeños en relación con la función de distribución de Maxwell. f gases con interna grados de libertad de simetría la conductividad térmica de un líquido, se puede utilizar una función de distribución de una sola partícula en equilibrio local con una sustancia química t-roy. potenciales e hidrodinámicos. velocidad, que corresponden al pequeño volumen de líquido considerado. Puede encontrar una corrección que sea proporcional a los gradientes del t-ry, hidrodinámico. velocidad y química potenciales de los componentes, y calcular los flujos de impulsos, energía y sustancias, así como justificar la ecuación de Navier-Stokes, la conductividad térmica y la difusión. En este caso, el coeficiente las transferencias resultan ser proporcionales a las correlaciones espacio-temporales. funciones de los flujos de energía, impulsos y sustancias de cada componente. Para describir los procesos de transferencia de material en sólidos y en las interfaces con un sólido, se utiliza ampliamente el modelo de condensador reticular. fases. Se describe principalmente la evolución del estado del sistema. cinético ecuación maestra sobre la función de distribución P(q, t):< N y),
q- номер узла или его координата. В модели "решеточного газа "
частица может находиться в узле (узел занят) или отсутствовать (узел свободен);
donde P(q,t)= t f(p,q,t)du- función de distribución, promediada sobre los impulsos (velocidades) de todas las N partículas, que describe la distribución de partículas sobre los nodos de la estructura reticular (su número es N y, N W(q : q") es la probabilidad de que el sistema pase por unidad de tiempo del estado q, descrito por un conjunto completo de coordenadas de partículas, a otro estado q". La primera suma describe la contribución de todos los procesos en los que se lleva a cabo una transición a un estado dado q, la segunda suma describe la salida de este estado. En el caso de distribución de partículas en equilibrio (t : , ) P(q) = exp[-H(q)/kT]/Q, donde Q-estadístico. En suma, H(q) es la energía del sistema en el estado q. Las probabilidades de transición satisfacen el principio de equilibrio detallado: Para calcular el coeficiente. transferencia en fases gaseosa, líquida y sólida, así como en los límites de fases, se utilizan activamente varias variantes del método mol. dinámica, que nos permite seguir en detalle la evolución del sistema desde tiempos ~10 -15 s a ~10 -10 s (en tiempos del orden de 10 -10 - 10 -9 s y más, el llamado Langevin Se utiliza una ecuación, esta ecuación contiene conceptos de Newton que contienen un término estocástico en el lado derecho). Para sistemas con químicos p-ciones, la naturaleza de la distribución de las partículas está muy influenciada por la relación entre los tiempos característicos de transferencia de los reactivos y sus propiedades químicas. transformaciones. Si la velocidad de la sustancia química La transformación es pequeña, la distribución de partículas no difiere mucho del caso cuando no hay solución. Si la velocidad de distribución es alta, su influencia sobre la naturaleza de la distribución de partículas es grande y es imposible utilizar concentraciones promedio de partículas (es decir, funciones de distribución con n = 1), como se hace cuando se utiliza la ley de acción de masas. Es necesario describir la distribución de reactivos con más detalle utilizando funciones de distribución f n con n > 1. Importante al describir reacciones. los flujos de partículas en la superficie y las velocidades de las reacciones controladas por difusión tienen condiciones límite (ver Macrocinética), 2ª ed., M., 1982; Curso de física de Berkeley, trad. Del inglés, 3ª ed., vol. 5-Reif F., Statistical Physics, M., 1986; Tovbin Yu.K., Teoría de los procesos físicos y químicos en la interfaz gas-sólido, M., 1990. Yu.K. Tovbin.
Física estadística y termodinámica.
Métodos de investigación estadística y termodinámica.
. La física molecular y la termodinámica son ramas de la física en las que se estudia. procesos macroscópicos en los cuerpos, asociado con la gran cantidad de átomos y moléculas contenidas en los cuerpos. Para estudiar estos procesos se utilizan dos métodos cualitativamente diferentes y mutuamente complementarios: estadístico (cinética molecular) Y termodinámico. El primero es la base de la física molecular, el segundo, la termodinámica. Física molecular
- una rama de la física que estudia la estructura y propiedades de la materia basándose en conceptos cinéticos moleculares, basándose en el hecho de que todos los cuerpos están formados por moléculas en continuo movimiento caótico. La idea de la estructura atómica de la materia fue expresada por el antiguo filósofo griego Demócrito (460-370 a. C.). El atomismo no volvió a resurgir hasta el siglo XVII. y se desarrolla en obras cuyas opiniones sobre la estructura de la materia y los fenómenos térmicos eran cercanas a las modernas. El desarrollo riguroso de la teoría molecular se remonta a mediados del siglo XIX. y está asociado con los trabajos del físico alemán R. Clausius (1822-1888), J. Maxwell y L. Boltzmann. Los procesos estudiados por la física molecular son el resultado de la acción combinada de una gran cantidad de moléculas. Las leyes de comportamiento de una gran cantidad de moléculas, al ser leyes estadísticas, se estudian utilizando método estadístico. Este método se basa en el hecho de que las propiedades de un sistema macroscópico están determinadas en última instancia por las propiedades de las partículas del sistema, las características de su movimiento y promediado valores de las características dinámicas de estas partículas (velocidad, energía, etc.). Por ejemplo, la temperatura de un cuerpo está determinada por la velocidad del movimiento caótico de sus moléculas, pero como en cualquier momento diferentes moléculas tienen diferentes velocidades, solo se puede expresar a través del valor promedio de la velocidad de movimiento de las moléculas. moléculas. No se puede hablar de la temperatura de una molécula. Por tanto, las características macroscópicas de los cuerpos tienen un significado físico sólo en el caso de un gran número de moléculas. Termodinámica- una rama de la física que estudia las propiedades generales de los sistemas macroscópicos en un estado de equilibrio termodinámico y los procesos de transición entre estos estados. La termodinámica no considera los microprocesos que subyacen a estas transformaciones. Este método termodinámico diferente al estadístico. La termodinámica se basa en dos principios: leyes fundamentales establecidas como resultado de la generalización de datos experimentales. El ámbito de aplicación de la termodinámica es mucho más amplio que el de la teoría cinética molecular, ya que no existen áreas de la física y la química en las que no se pueda utilizar el método termodinámico. Sin embargo, por otro lado, el método termodinámico es algo limitado: la termodinámica no dice nada sobre la estructura microscópica de la materia, sobre el mecanismo de los fenómenos, sino que sólo establece conexiones entre las propiedades macroscópicas de la materia. La teoría cinética molecular y la termodinámica se complementan, formando un todo único, pero diferenciándose en varios métodos de investigación. Postulados básicos de la teoría cinética molecular (MKT)
1.
Todos los cuerpos en la naturaleza están formados por una gran cantidad de partículas diminutas (átomos y moléculas). 2.
Estas partículas están en continuo caótico movimiento (desordenado). 3.
El movimiento de las partículas está relacionado con la temperatura corporal, por eso se le llama movimiento térmico.
4.
Las partículas interactúan entre sí. Evidencias de la validez de MCT: difusión de sustancias, movimiento browniano, conductividad térmica. Las cantidades físicas utilizadas para describir procesos en física molecular se dividen en dos clases: microparámetros– cantidades que describen el comportamiento de partículas individuales (masa de un átomo (molécula), velocidad, momento, energía cinética de partículas individuales); La temperatura es uno de los conceptos básicos que juega un papel importante no sólo en la termodinámica, sino también en la física en general. Temperatura- una cantidad física que caracteriza el estado de equilibrio termodinámico de un sistema macroscópico. De acuerdo con la decisión de la XI Conferencia General de Pesas y Medidas (1960), actualmente solo se pueden utilizar dos escalas de temperatura: termodinámico Y practica internacional, graduados respectivamente en kelvins (K) y grados Celsius (°C). En la escala termodinámica, el punto de congelación del agua es 273,15 K (al mismo presión como en la Escala Práctica Internacional), por lo tanto, por definición, temperatura termodinámica y Temperatura Práctica Internacional escala están relacionadas por la relación t= 273,15 +
t.
Temperatura t
= 0 K se llama cero kelvin. El análisis de varios procesos muestra que 0 K es inalcanzable, aunque es posible acercarse a él tanto como se desee. 0 K es la temperatura a la que teóricamente debería cesar todo movimiento térmico de las partículas de una sustancia. En física molecular se deriva una relación entre macroparámetros y microparámetros. Por ejemplo, la presión de un gas ideal se puede expresar mediante la fórmula: posición: relativa; arriba:5.0pt">- masa de una molécula, - concentración, font-size: 10.0pt">De la ecuación básica MKT puede obtener una ecuación conveniente para uso práctico: font-size: 10.0pt">Un gas ideal es un modelo de gas idealizado en el que se cree que: 1.
el volumen intrínseco de las moléculas de gas es insignificante en comparación con el volumen del recipiente; 2.
no existen fuerzas de interacción entre moléculas (atracción y repulsión a distancia; 3.
Las colisiones de moléculas entre sí y con las paredes del recipiente son absolutamente elásticas. Un gas ideal es un modelo teórico simplificado de un gas. Sin embargo, esta ecuación puede describir el estado de muchos gases bajo ciertas condiciones. Para describir el estado de los gases reales, se deben introducir correcciones en la ecuación de estado. La presencia de fuerzas repulsivas que contrarrestan la penetración de otras moléculas en el volumen ocupado por una molécula significa que el volumen libre real en el que pueden moverse las moléculas de un gas real será menor. Dóndeb -
el volumen molar ocupado por las propias moléculas. La acción de las fuerzas de atracción del gas provoca la aparición de una presión adicional sobre el gas, denominada presión interna. Según los cálculos de van der Waals, la presión interna es inversamente proporcional al cuadrado del volumen molar, es decir, donde A - Constante de van der Waals, que caracteriza las fuerzas de atracción intermolecular,V metro -
Volumen molar. Al final lo conseguiremos ecuación de estado del gas real o ecuación de van der Waals:
font-size:10.0pt;font-family:" times new roman> Significado físico de temperatura: la temperatura es una medida de la intensidad del movimiento térmico de partículas de sustancias. El concepto de temperatura no es aplicable a una molécula individual. Sólo para un número suficientemente grande de moléculas que crean una cierta cantidad de sustancia, tiene sentido incluir el término temperatura. Para un gas monoatómico ideal, podemos escribir la ecuación: font-size:10.0pt;font-family:" times new roman>La primera determinación experimental de las velocidades moleculares fue realizada por el físico alemán O. Stern (1888-1970). Sus experimentos también permitieron estimar la distribución de las velocidades. de moléculas. El "enfrentamiento" entre las energías potenciales de unión de las moléculas y las energías del movimiento térmico de las moléculas (moléculas cinéticas) conduce a la existencia de varios estados agregados de la materia. Termodinámica Contando el número de moléculas en un sistema dado y estimando sus energías cinética y potencial promedio, podemos estimar la energía interna de un sistema dado. Ud. font-size:10.0pt;font-family:" times new roman>Para un gas monoatómico ideal. La energía interna de un sistema puede cambiar como resultado de varios procesos, por ejemplo, realizar trabajo en el sistema o impartirle calor. Entonces, al empujar un pistón dentro de un cilindro que contiene gas, comprimimos este gas, como resultado de lo cual su temperatura aumenta, es decir, cambiando (aumentando) la energía interna del gas. Por otro lado, la temperatura de un gas y su energía interna se pueden aumentar impartiéndole una cierta cantidad de calor: energía transferida al sistema por cuerpos externos a través del intercambio de calor (el proceso de intercambio de energías internas cuando los cuerpos entran en contacto). con diferentes temperaturas). Así, podemos hablar de dos formas de transferencia de energía de un cuerpo a otro: trabajo y calor. La energía del movimiento mecánico se puede convertir en energía del movimiento térmico y viceversa. Durante estas transformaciones se observa la ley de conservación y transformación de la energía; En relación con los procesos termodinámicos, esta ley es primera ley de la termodinámica, establecido como resultado de la generalización de datos experimentales centenarios: Por lo tanto, en un circuito cerrado font-size:10.0pt;font-family:" times new roman>Eficiencia del motor térmico: De la primera ley de la termodinámica se deduce que la eficiencia de un motor térmico no puede ser superior al 100%. Al postular la existencia de diversas formas de energía y la conexión entre ellas, el primer principio de TD no dice nada sobre la dirección de los procesos en la naturaleza. En total conformidad con el primer principio, se puede construir mentalmente un motor en el que se realice un trabajo útil reduciendo la energía interna de la sustancia. Por ejemplo, en lugar de combustible, un motor térmico usaría agua y, enfriando el agua y convirtiéndola en hielo, se realizaría trabajo. Pero estos procesos espontáneos no ocurren en la naturaleza. Todos los procesos de la naturaleza se pueden dividir en reversibles e irreversibles. Durante mucho tiempo, uno de los principales problemas de las ciencias naturales clásicas siguió siendo el problema de explicar la naturaleza física de la irreversibilidad de los procesos reales. La esencia del problema es que el movimiento de un punto material, descrito por la ley II de Newton (F = ma), es reversible, mientras que un gran número de puntos materiales se comportan de forma irreversible. Si el número de partículas en estudio es pequeño (por ejemplo, dos partículas en la figura a)), entonces no podremos determinar si el eje del tiempo está dirigido de izquierda a derecha o de derecha a izquierda, ya que cualquier secuencia de fotogramas es igualmente posible. esto es todo fenómeno reversible.
La situación cambia significativamente si el número de partículas es muy grande (Fig. b)). En este caso, la dirección del tiempo se determina de manera inequívoca: de izquierda a derecha, ya que es imposible imaginar que las partículas distribuidas uniformemente por sí solas, sin influencias externas, se reúnan en la esquina de la "caja". Este comportamiento, cuando el estado del sistema solo puede cambiar en una secuencia determinada, se llama irreversible. Todos los procesos reales son irreversibles. Ejemplos de procesos irreversibles: difusión, conductividad térmica, flujo viscoso. Casi todos los procesos reales en la naturaleza son irreversibles: como la amortiguación de un péndulo, la evolución de una estrella y la vida humana. La irreversibilidad de los procesos en la naturaleza, por así decirlo, marca la dirección en el eje del tiempo desde el pasado hacia el futuro. El físico y astrónomo inglés A. Eddington llamó en sentido figurado a esta propiedad del tiempo "la flecha del tiempo". ¿Por qué, a pesar de la reversibilidad del comportamiento de una partícula, un conjunto de un gran número de tales partículas se comporta de manera irreversible? ¿Cuál es la naturaleza de la irreversibilidad? ¿Cómo justificar la irreversibilidad de los procesos reales basándose en las leyes de la mecánica de Newton? Estas y otras cuestiones similares preocuparon a los científicos más destacados de los siglos XVIII y XIX. Segunda ley de la termodinámica
establece la dirección pereza de todos los procesos en sistemas aislados. Aunque se conserva la cantidad total de energía en un sistema aislado, su composición cualitativa cambia irreversiblemente.
1.
En la formulación de Kelvin, la segunda ley es: "No existe ningún proceso posible cuyo único resultado sea la absorción de calor de un calentador y la conversión completa de este calor en trabajo". 2.
En otra formulación: “El calor sólo puede transferirse espontáneamente de un cuerpo más calentado a uno menos calentado”. 3.
La tercera formulación: "La entropía en un sistema cerrado sólo puede aumentar". Segunda ley de la termodinámica prohíbe la existencia máquina de movimiento perpetuo del segundo tipo
,
es decir, una máquina capaz de realizar un trabajo transfiriendo calor de un cuerpo frío a uno caliente. La segunda ley de la termodinámica indica la existencia de dos formas diferentes de energía: el calor como medida del movimiento caótico de partículas y el trabajo asociado con el movimiento ordenado. El trabajo siempre se puede convertir en su equivalente calor, pero el calor no se puede convertir completamente en trabajo. Por tanto, una forma desordenada de energía no puede transformarse en ordenada sin acciones adicionales. Completamos la transformación del trabajo mecánico en calor cada vez que pisamos el pedal del freno de un coche. Pero sin ninguna acción adicional en el ciclo cerrado de funcionamiento del motor, es imposible transferir todo el calor al trabajo. Parte de la energía térmica se gasta inevitablemente en calentar el motor, además el pistón en movimiento trabaja constantemente contra las fuerzas de fricción (esto también consume energía mecánica). Pero el significado de la segunda ley de la termodinámica resultó ser aún más profundo. Otra formulación de la segunda ley de la termodinámica es la siguiente afirmación: la entropía de un sistema cerrado es una función no decreciente, es decir, durante cualquier proceso real aumenta o permanece sin cambios. El concepto de entropía, introducido en la termodinámica por R. Clausius, fue inicialmente artificial. El destacado científico francés A. Poincaré escribió sobre esto: “La entropía parece algo misteriosa en el sentido de que esta cantidad es inaccesible a cualquiera de nuestros sentidos, aunque tiene la propiedad real de las cantidades físicas, ya que, al menos en principio, es completamente mensurable " Según la definición de Clausius, la entropía es una cantidad física cuyo incremento es igual a la cantidad de calor.
, recibido por el sistema, dividido por la temperatura absoluta: font-size:10.0pt;font-family:" times new roman>De acuerdo con la segunda ley de la termodinámica, en sistemas aislados, es decir, sistemas que no intercambian energía con el medio ambiente, un estado desordenado (caos) no puede transformarse de forma independiente en orden Así, en sistemas aislados, la entropía sólo puede aumentar. principio de entropía creciente. Según este principio, cualquier sistema tiende a alcanzar un estado de equilibrio termodinámico, que se identifica con el caos. Dado que un aumento de la entropía caracteriza los cambios a lo largo del tiempo en sistemas cerrados, la entropía actúa como una especie de flechas del tiempo.
Llamamos desordenado al estado con entropía máxima y ordenado al estado con entropía baja. Un sistema estadístico, si se deja a su suerte, pasa de un estado ordenado a uno desordenado con una entropía máxima correspondiente a parámetros externos e internos dados (presión, volumen, temperatura, número de partículas, etc.). Ludwig Boltzmann relacionó el concepto de entropía con el concepto de probabilidad termodinámica: font-size:10.0pt;font-family:" times new roman> Así, cualquier sistema aislado, abandonado a su suerte, con el tiempo pasa de un estado de orden a un estado de máximo desorden (caos). De este principio se sigue una hipótesis pesimista sobre muerte por calor del universo, formulado por R. Clausius y W. Kelvin, según el cual: ·
la energía del Universo es siempre constante; ·
La entropía del Universo siempre está aumentando. Así, todos los procesos del Universo tienen como objetivo alcanzar un estado de equilibrio termodinámico, correspondiente al estado de mayor caos y desorganización. Todos los tipos de energía se degradan, convirtiéndose en calor, y las estrellas pondrán fin a su existencia, liberando energía al espacio circundante. Una temperatura constante se establecerá sólo unos pocos grados por encima del cero absoluto. En este espacio habrá planetas y estrellas enfriados y sin vida. No habrá nada: ni fuentes de energía, ni vida. Esta sombría perspectiva fue predicha por la física hasta la década de 1960, aunque las conclusiones de la termodinámica contradecían los resultados de la investigación en biología y ciencias sociales. Así, la teoría evolutiva de Darwin atestigua que la naturaleza viva se desarrolla principalmente en la dirección de la mejora y complejidad de nuevas especies de plantas y animales. La historia, la sociología, la economía y otras ciencias sociales y humanas también han demostrado que en la sociedad, a pesar de los zigzagueos individuales del desarrollo, en general se observa progreso. La experiencia y la actividad práctica han demostrado que el concepto de sistema cerrado o aislado es una abstracción bastante burda que simplifica la realidad, ya que en la naturaleza es difícil encontrar sistemas que no interactúen con el medio ambiente. La contradicción comenzó a resolverse cuando en termodinámica, en lugar del concepto de sistema cerrado y aislado, se introdujo el concepto fundamental de sistema abierto, es decir, un sistema que intercambia materia, energía e información con el medio ambiente.![]() desde probabilidad pmín encontrar una partícula en una celda dada es igual a V 0
/
V. Si en momentos posteriores la partícula comienza a llenar un volumen mayor, se perderá información y aumentará la entropía. Enfatizamos que es necesario pagar por la información (según la segunda ley) aumentando la entropía. se sistema externo, y de hecho, si por un bit de información el dispositivo (sistema externo) aumentara su entropía en una cantidad menor a un bit, entonces podríamos revertir el motor térmico. Es decir, al expandir el volumen ocupado por una partícula, aumentaríamos su entropía en la cantidad en2
conseguir un trabajo Tln2
, y la entropía total del sistema partícula más dispositivo disminuiría. Pero esto es imposible según el segundo principio. Formalmente,
desde probabilidad pmín encontrar una partícula en una celda dada es igual a V 0
/
V. Si en momentos posteriores la partícula comienza a llenar un volumen mayor, se perderá información y aumentará la entropía. Enfatizamos que es necesario pagar por la información (según la segunda ley) aumentando la entropía. se sistema externo, y de hecho, si por un bit de información el dispositivo (sistema externo) aumentara su entropía en una cantidad menor a un bit, entonces podríamos revertir el motor térmico. Es decir, al expandir el volumen ocupado por una partícula, aumentaríamos su entropía en la cantidad en2
conseguir un trabajo Tln2
, y la entropía total del sistema partícula más dispositivo disminuiría. Pero esto es imposible según el segundo principio. Formalmente, ![]() Por tanto, una disminución de la entropía del sistema (partícula) va acompañada de un aumento de la entropía del dispositivo.
Por tanto, una disminución de la entropía del sistema (partícula) va acompañada de un aumento de la entropía del dispositivo.![]() , donde (esto se refiere a sistemas de dos niveles, como bits: “0” y “1”. Si la dimensión es norte, Eso h =
iniciar sesión sustantivo, masculino—.
Si, por norte
= 3, norte =registro 3
y, = 3.)
, donde (esto se refiere a sistemas de dos niveles, como bits: “0” y “1”. Si la dimensión es norte, Eso h =
iniciar sesión sustantivo, masculino—.
Si, por norte
= 3, norte =registro 3
y, = 3.)![]()
![]()
![]()
![]() moléculas donde v i son las frecuencias de las normas
moléculas donde v i son las frecuencias de las normas ![]()
parámetros macro– cantidades que no pueden reducirse a partículas individuales, pero que caracterizan las propiedades de la sustancia en su conjunto. Los valores de los macroparámetros están determinados por el resultado de la acción simultánea de una gran cantidad de partículas. Los parámetros macro son temperatura, presión, concentración, etc.![]() .
.

Material de FFWiki.
| Artículo | Termodinámica y física estadística. | Semestre | 7-8 | Tipo | conferencia, seminario | Informes | examen | Departamento | Departamento de Estadística Cuántica y Teoría de Campos |
|---|
Sobre el artículo
Termodinámica y física estadística. La primera pregunta cuando ves este tema en la agenda es: ¿cómo es posible? De hecho, en el primer año ya enseñaban física molecular, que incluía los 3 principios de la termodinámica, los potenciales y la distribución de Maxwell. Al parecer, ¿qué más podría haber de nuevo en la naturaleza?
Resulta que lo que había en el primer año es charla infantil en comparación con la termodinámica real y la física estadística. Aquel con el que Landau calculó el helio líquido y recibió el Premio Nobel.
Es importante no caer en la trampa de pensar que sólo porque en una conferencia te cuentan lo que sabías en la escuela, entonces seguirá siendo así. Ya a partir de mediados de septiembre serán testigos de sorprendentes trucos con derivadas parciales y, a finales del semestre de otoño, habrá temas muy espeluznantes en física estadística:
- Cálculo de sumas estadísticas y distribuciones de Gibbs.
- Gases cuánticos: gases Fermi y Bose en diferentes condiciones
- Transiciones de fase y sus propiedades.
- Gases no ideales: cadenas de Bogolyubov, modelos de plasma y electrolitos.
El autor de estas palabras, aunque supo prepararse perfectamente 4 días antes de los exámenes, está muy arrepentido de ello y no aconseja a nadie que repita semejante violencia contra su cerebro :) Las tareas y preguntas del examen se conocen desde el principios de año y es muy útil preparar parte del material con antelación.
Hay temas tanto fáciles como difíciles en el semestre de primavera. Por ejemplo, la teoría del movimiento browniano es muy fácil de escribir. Pero al final del curso hay varias ecuaciones cinéticas, que son mucho más difíciles de entender.
Examen
El examen de otoño va bastante bien, realmente no te permiten hacer trampa. En su mayor parte, los profesores no se portan mal, pero tampoco ha habido obsequios notables. Necesitas conocer la teoría. El diploma incluye la evaluación para el examen de primavera. El examen de primavera es más difícil en términos de material que el examen de otoño, pero normalmente se acepta con mayor capacidad de respuesta. Sin embargo, la teoría también debe conocerse bien.
El billete tanto para otoño como para primavera contiene 2 preguntas teóricas y una tarea.
Tenga cuidado con sus estadísticas, varias personas (¡el número varía de 2 a 10!) regularmente se gradúan sin aprobar este examen. Y estos no son cualquiera, sino curtidos estudiantes de cuarto año.
Materiales
semestre de otoño
- Folleto 7 semestre (pdf) - folleto útil
- Respuestas a preguntas sobre termodinámica (pdf) - 1 pregunta por entrada
- Respuestas a preguntas sobre física estadística (pdf) - pregunta 2 del billete
- https://vk.com/doc231234703_450962027?hash=b2eb6c2220f95a7795&dl=cf7b5295f12e0fd4e0 tareas para el primer coloquio
- https://vk.com/doc181113102_453848269?hash=6d6f68abc3358e2adf&dl=1606060abdd44d226e al segundo
semestre de primavera
- Respuestas a las preguntas del examen, teoría (pdf): respuestas a las preguntas del examen teórico cuidadosamente escritas en computadoras.
- - resolución de problemas
- Soluciones a problemas para el examen (pdf) - más soluciones a problemas
Literatura
Libros de problemas
- Tareas sobre termodinámica y física estadística para estudiantes de cuarto año de la Facultad de Física de la Universidad Estatal de Moscú (semestre de otoño - teoría de sistemas de equilibrio) (pdf)